pkgs <- c("fs", "futile.logger", "configr", "stringr", "ggpubr", "jhtools",
"glue", "ggsci", "patchwork", "tidyverse", "ggthemes","tidyplots")
for (pkg in pkgs){
suppressPackageStartupMessages(library(pkg, character.only = T))
}
# set workdir
setwd("/cluster/home/yjliu_jh/projects/mm/output/igdlike2")
# select altered genes
mm1199_full <- read_rds("/cluster/home/jhuang/projects/mm/analysis/meta/human/rnaseq/exp/mm1199_full.rds")
exp_mm <- mm1199_full[, -2] %>% left_join(hugo_anno) %>% na.omit() %>%
dplyr::select(-c(gene_id, locus_group)) %>% as.data.frame() %>% remove_rownames() %>%
column_to_rownames(var = "symbol") %>% as.matrix()
up_example_genes <- c("MYH10", "IGHD")
down_example_genes <- c("KIT", "FAT4", "ETV1", "CDH2", "NRG3", "NDNF")
selected_exp <- exp_mm[rownames(exp_mm) %in% c(up_example_genes, down_example_genes), ]
long_exp <- selected_exp %>% as.data.frame() %>% rownames_to_column(var = "gene") %>%
pivot_longer(-gene, names_to = "sample_id", values_to = "value")
merged_sinfo <- read_rds("~/projects/mm/docs/meta/sampleinfo/sampleinfo_jilin_commpass.rds")
sinfo_part <- merged_sinfo[, c("sample_id", "subtypes", "batch")] %>% na.omit() %>% unique()
long_exp <- left_join(long_exp, sinfo_part)
long_exp <- na.omit(long_exp)
# get color
config_fn = "~/projects/mm/analysis/jilin/human/rnaseq/configs/colors.yaml"
config_list <- show_me_the_colors(config_fn, "all")
group_colors <- c(config_list[["batch"]][1:2], "public" = "#BBA343")
leo <- long_exp[order(long_exp$subtypes), ]
# exp plot
for (gene in c(up_example_genes, down_example_genes)) {
col_ordered <- group_colors[leo[leo$gene %in% gene, ]$batch]
legend_df <- data.frame(label = names(group_colors), x = 1:3, y = 1)
legend_plot <- ggplot(legend_df, aes(x = x, y = x, color = label)) +
geom_point(size = 4) +
scale_color_manual(values = group_colors, name = "Batch") +
theme_void() + theme(legend.position = "right")
legend_grob <- cowplot::get_legend(legend_plot)
main_plot <- long_exp[long_exp$gene %in% gene, ] |>
tidyplot(x = subtypes, y = value, color = subtypes) |>
add_violin() |> adjust_size(width = 100, height = 60) |>
tidyplots::add_data_points_beeswarm(col = col_ordered, size = 0.3) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
ggtitle(glue("{gene}"))
final_plot <- cowplot::plot_grid(main_plot, legend_grob, rel_widths = c(1, 0.2))
pdf(glue("exp_plots/{gene}_subtypes.pdf"), width = 7, height = 5)
print(final_plot)
dev.off()
}16 A specific transcriptional subtype
During the clustering process, we observed that certain genomic or clinical features exhibited strong concordance with transcriptomic profiles, as samples harboring these features consistently formed distinct clusters in unsupervised analyses. Following robustness validation, we identified one such cluster as a new molecular subtype, designated the IgD-enriched subtype.
Multiple myeloma (MM) can be classified based on the type of immunoglobulin (Ig) secreted by malignant plasma cells. Among these, the IgD subtype is relatively rare and has been previously associated with adverse clinical features such as amyloidosis. Prior studies [PMID: 33082513] have reported a higher prevalence of IgD-type MM in Asian populations, consistent with our observations. Importantly, we first observed that specific transcriptomic features could be related to the IgD subtype, highlighting its molecular distinctiveness.
After expression values were adjusted to account for batch effects, we found the genes highly expressed in the IgD group based were no longer significantly higher than other groups, suggesting potential residual technical influence. In contrast, downregulated genes in this group have mostly kept the same, likely reflect biologically meaningful characteristics.
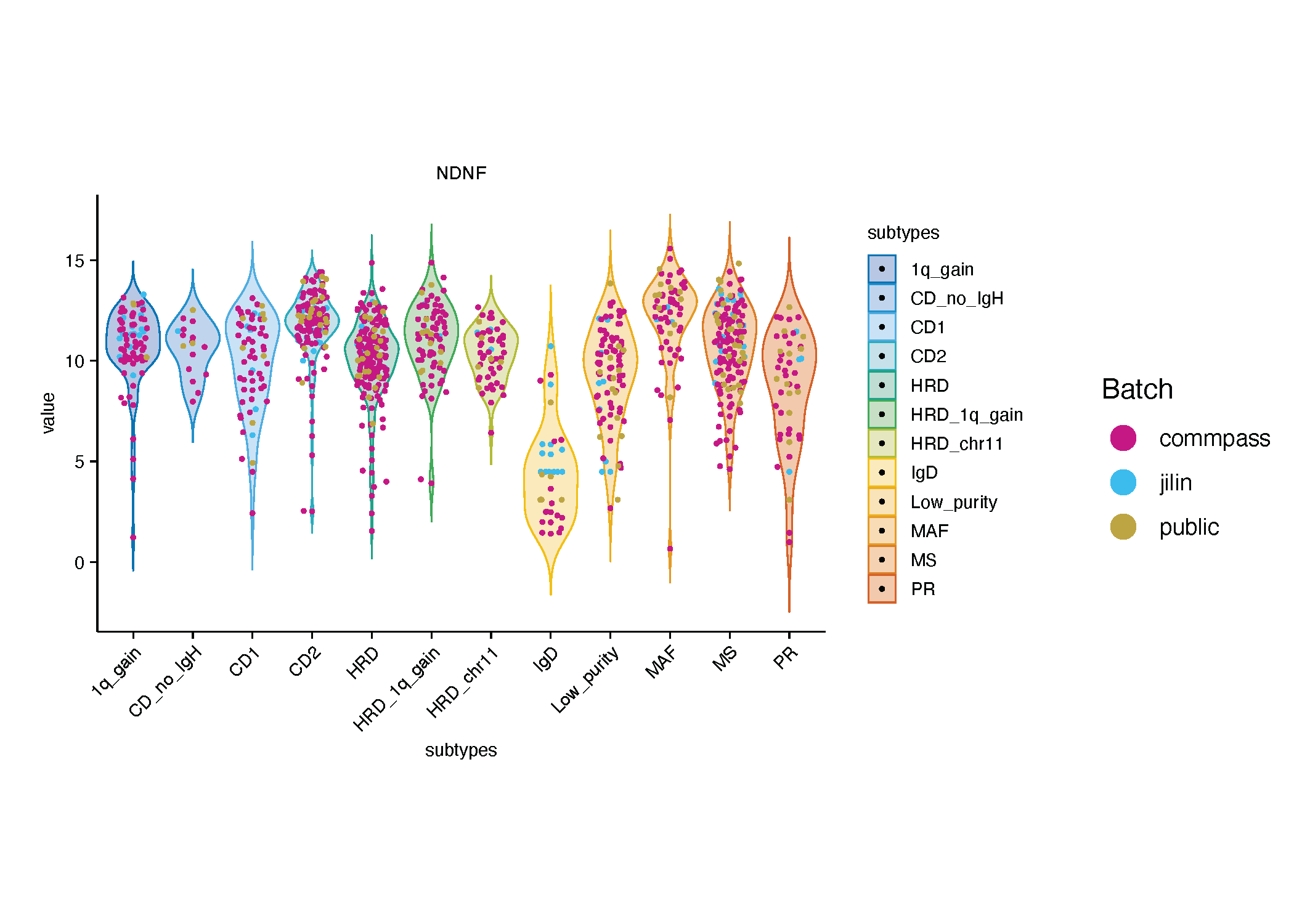
These downregulated genes included canonical signaling and adhesion molecules such as KIT, FAT4, ETV1, and CDH2, as well as less well-studied candidates like NRG3 and NDNF. Collectively, these transcriptional alterations suggest that the IgD-enriched subtype represents a biologically distinct entity within MM, characterized by a molecular program that is skewed away from immune activation and toward chromatin remodeling, epithelial architecture, and reduced immunoglobulin-mediated signaling.