# Load necessary R packages for data processing and visualization
pkgs <- c("fs", "futile.logger", "configr", "ggpubr", "ggthemes", "jhtools",
"glue", "ggsci", "patchwork", "tidyverse", "circlize", "ggrepel",
"ComplexHeatmap", "GenomicRanges", "jhuanglabRNAseq", "ggh4x")
for (pkg in pkgs){
suppressPackageStartupMessages(library(pkg, character.only = T))
}
# Define project parameters
project <- "mm"
dataset <- "meta"
species <- "human"
workdir <- glue("~/projects/{project}/output/{dataset}/{species}/figures/fig1") |> checkdir()
setwd(workdir)31 Figure 1
31.1 Figure 1
31.2 Setup
Load required R packages and set the working directory.
31.3 enrich score of public gene sets in MM subtypes
# read in data
objht <- read_rds("/cluster/home/jhuang/projects/mm/analysis/meta/human/rnaseq/figures/heatmap/step1/heatmap_0.9_groups16.rds")
df <- read_rds("/cluster/home/jhuang/projects/mm/docs/meta/sampleinfo/sampleinfo_jilin_commpass.rds")
config_fn <- "~/projects/mm/output/meta/human/figures/colors_fix.yaml"
config_list <- show_me_the_colors(config_fn, "all")
subdf <- tibble(sample_id = rownames(objht$heatmap2[[6]])) |>
left_join(df) |>
mutate(
treatment_group = case_when(treatment_group %in% "Unknown" ~ NA_character_,
T ~ treatment_group),
hit_event = str_extract(hit_event, pattern = "\\d+"),
best_response = case_when(
best_response == "SCR" ~ "sCR",
best_response == "≥VGPR" ~ "VGPR",
best_response == "Unknown" ~ NA_character_,
T ~ best_response
)
)
out_dir <- dir_create("./score_dotplot/")
all_scores <- c("subtypes", config_list$paper_scores)
subdf_score <- subdf[,all_scores]
subdf_score[,config_list$paper_scores] <- apply(subdf_score[,config_list$paper_scores], 2, function(x){
(x-min(x))/(max(x) - min(x))
# scale(x)
})
subdf_score$subtypes = factor(subdf_score$subtypes, levels = rev(unique(subdf_score$subtypes)))
dotplotdf <- subdf_score |>
pivot_longer(- subtypes) |>
group_by(subtypes, name) |>
summarise(
positive_rate = sum(value > 0.5)/n(),
mean_score = mean(value)) |>
ungroup() |>
mutate(name = factor(name, levels = config_list$paper_scores))
score_dotplot <- ggplot(dotplotdf |> mutate(label = str_extract(name, pattern = ".+?et_al")),
aes(x = name, y = subtypes, fill = mean_score, size = positive_rate)) +
geom_point(shape = 21)+
scale_fill_gradientn(limits = c(0.1, 0.9),
colours = c("#1E90FF", "white","#DC143C"),
values = scales::rescale(c(0.1, 0.5, 0.9)),
oob = scales::squish,
name = "Z-Score")+
theme_few() +
facet_grid(1~ label, scales = "free_x", space = "free_x") +
theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1),
axis.title.x = element_blank(),
# axis.text.y = element_blank(),
axis.ticks.length.y = unit(0, "mm"),
axis.ticks.length.x = unit(0, "mm"),
legend.position = "right",
panel.spacing = unit(-1, "mm"),
strip.text.y.right = element_blank(),
strip.text.x.top = element_blank(),
strip.background = element_rect(fill = NA),
# plot.margin = margin(l = 1),
panel.border = element_rect(fill = NA, color = "black", size = 0.5))
pdf(glue("{out_dir}/score_dotplot.pdf"), width = 12, height = 5)
print(score_dotplot)
dev.off()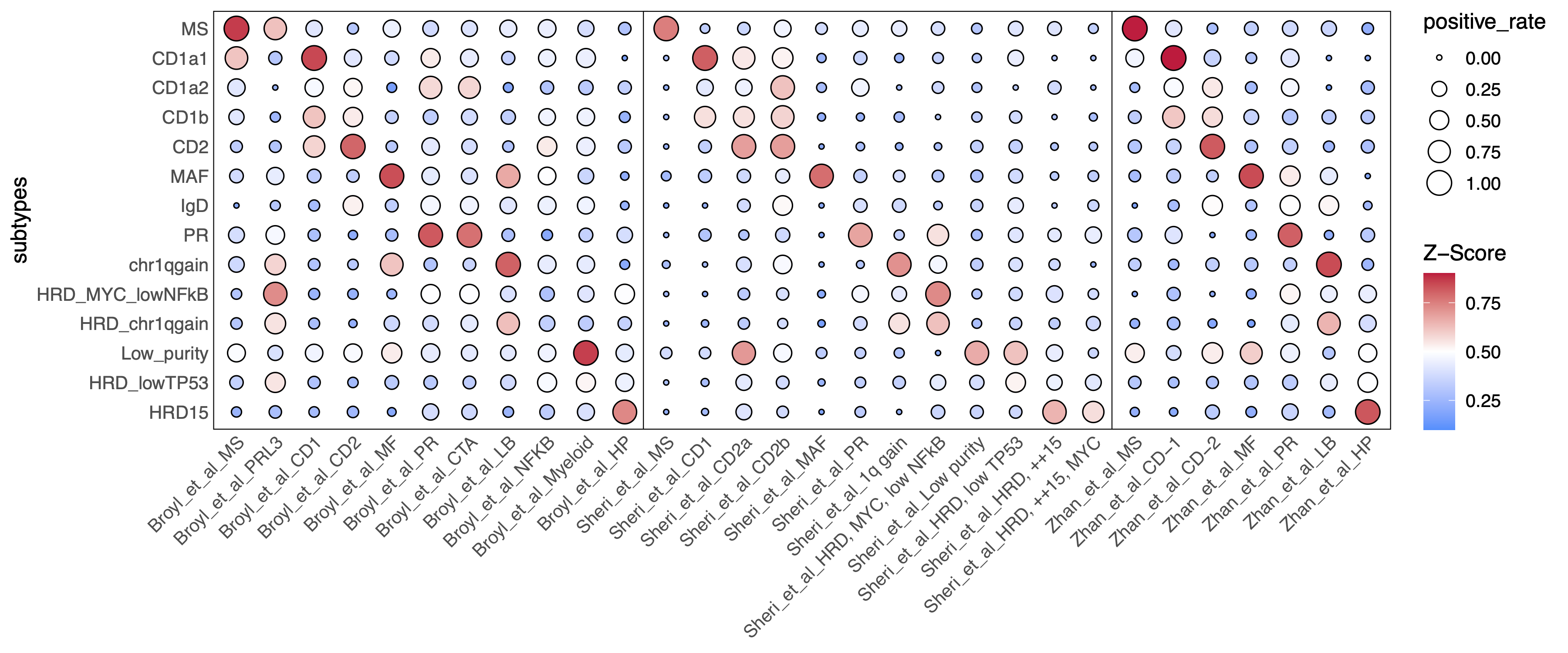
31.4 Refined molecular subtypes of MM
# colors settings
col <- config_list[c("gender", "ethnicity", "age", "GEP70", "Clinical_IgH", "Clinical_IgL", "batch", "subtypes", "imwg_risk",
"treatment_group", "best_response", "hit_event")]
col$ethnicity <- c(col$ethnicity, "other" = "grey")
col$datasets <- config_list$batch
col$GEP70 <- colorRamp2(quantile(subdf$GEP70, c(0.1, 0.5, 0.9), na.rm = TRUE),
colors = col$GEP70[c("colors1", "colors2", "colors3")])
col$age <- colorRamp2(quantile(subdf$age, as.numeric(col$age[c("breaks1", "breaks2", "breaks3")]), na.rm = TRUE),
colors = col$age[c("colors1", "colors2", "colors3")])
out_dir <- dir_create("./cluster_heatmap/")
# draw
all_col <- config_list$clinical_order
show_legend <- config_list$show_legend
hasub2 <- HeatmapAnnotation(df = subdf[,all_col],
annotation_name_side = "left",
show_legend = all_col %in% show_legend,
col = col)
ht <- Heatmap(t(objht$heatmap2[[6]]),
show_column_names = FALSE,
show_row_names = FALSE,
top_annotation = hasub2,
name = "Z-Score",
border = TRUE,
cluster_columns = FALSE,
cluster_rows = FALSE,
column_gap = unit(0, "mm"),
column_title_rot = 90,
clustering_method_columns = "ward.D2",
clustering_method_rows = "ward.D2",
heatmap_width = unit(160, "mm"),
column_split = as.numeric(factor(subdf$subtypes, levels = unique(subdf$subtypes))),
column_title = unique(subdf$subtypes))
pdf(glue("{out_dir}/figure1_heatmap.pdf"), width = 14, height = 6)
draw(ht, annotation_legend_side = "right", heatmap_legend_side = "left")
dev.off()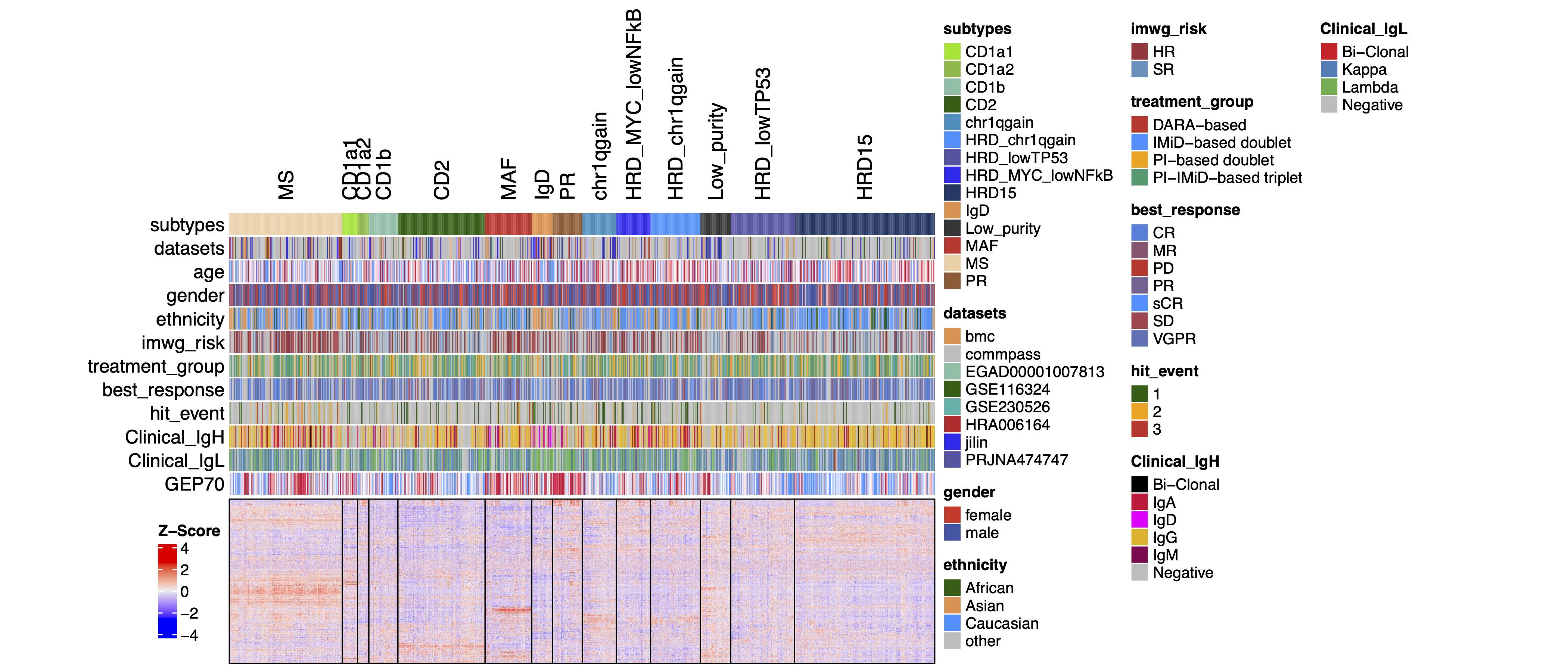
31.5 TSNE of subtypes
out_dir <- dir_create("./tsne/")
mm_heatmap <- "/cluster/home/jhuang/projects/mm/analysis/meta/human/rnaseq/exp/mm_heatmap1117.rds" %>%
read_rds() |> convert_df_plot()
dat_exp <- mm_heatmap[colnames(objht$heatmap2[[6]]), ] |> t()
pca_raw <- prcomp(dat_exp, scale. = TRUE)
pca_mat <- as.data.frame(pca_raw$x[, 1:2])
tsne_raw <- Rtsne::Rtsne(dat_exp, perplexity = 30, max_iter = 1000,
verbose = FALSE, check_duplicates = FALSE)
tsne_mat <- as.data.frame(tsne_raw$Y)
colnames(tsne_mat) <- c("tSNE_1", "tSNE_2")
rownames(tsne_mat) <- rownames(dat_exp)
tsne_mat$sample_id <- rownames(tsne_mat)
tsne_mat <- tsne_mat |>
left_join(
subdf[,c("sample_id", "subtypes", "datasets", "ethnicity", "Clinical_IgH")] |>
mutate(
datasets = factor(datasets, levels = c("HRA006164", "jilin", "PRJNA474747", "bmc", "GSE116324", "GSE230526", "EGAD00001007813", "commpass")),
ethnicity = factor(ethnicity, levels = c("Asian", "African", "Caucasian", "other")),
Clinical_IgH = factor(Clinical_IgH, levels = c("IgM", "Bi-Clonal", "IgD", "IgA", "IgG", "Negative")),
)
)
pdf(glue("{out_dir}/embedding_plot.pdf"), width = 7, height = 6)
ggplot(tsne_mat,
aes(x = tSNE_1, y = tSNE_2, color = subtypes)) +
geom_point() +
ggrepel::geom_label_repel(data = tsne_mat |>
group_by(subtypes) |>
summarise(tSNE_1 = mean(tSNE_1),
tSNE_2 = mean(tSNE_2)),
mapping = aes(x = tSNE_1, y = tSNE_2, label = subtypes, fill = subtypes), color = "white") +
scale_color_manual(values = col$subtypes) +
scale_fill_manual(values = col$subtypes) +
theme_void() +
theme(legend.position = "none")
dev.off()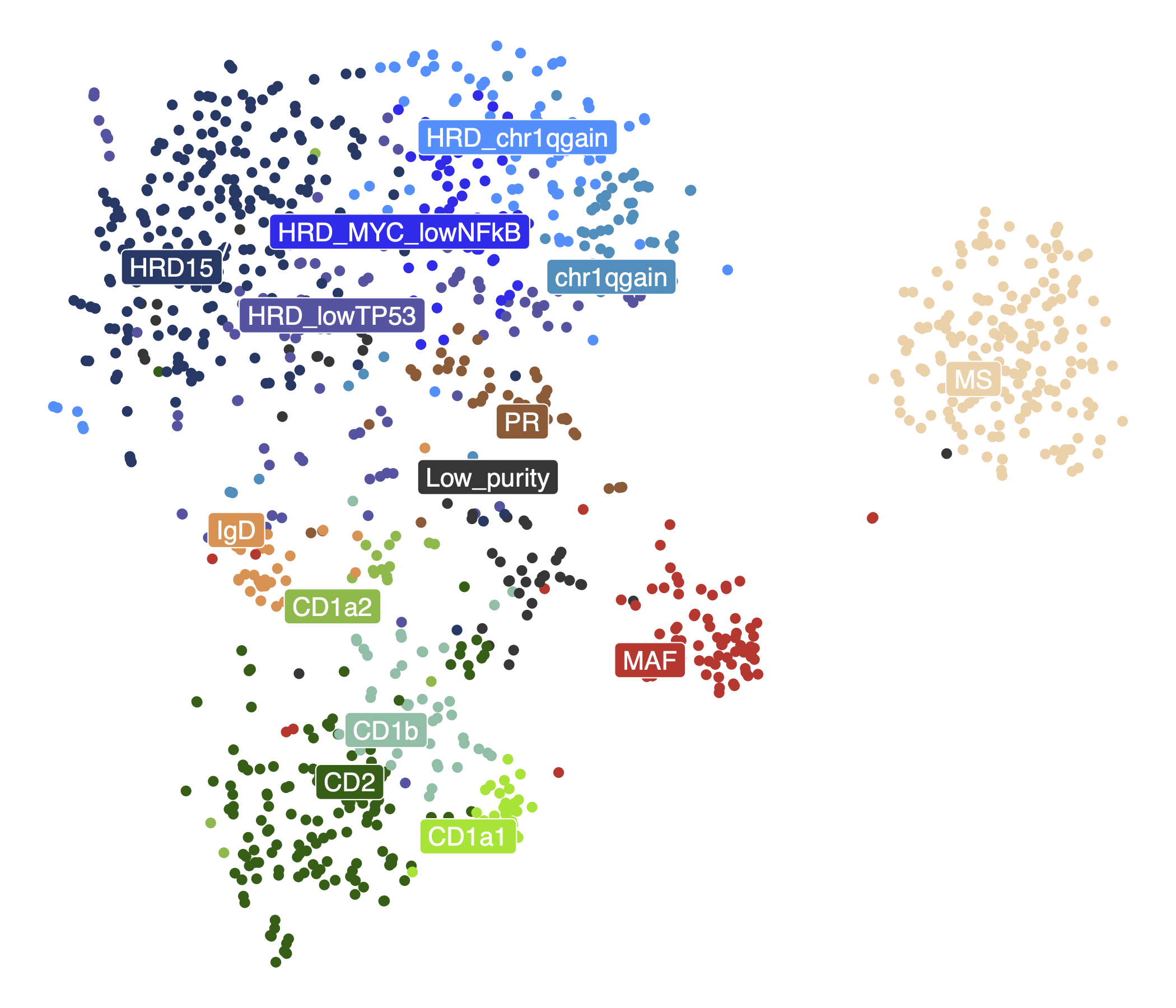
31.5.1 clinical features of subtypes
code: