pkgs <- c("fs", "futile.logger", "configr", "stringr", "ggpubr", "ggthemes",
"jhtools", "glue", "ggsci", "patchwork", "tidyverse", "dplyr",
"SummarizedExperiment", "jhuanglabRNAseq", "DESeq2", "gplots",
"matrixStats", "Matrix", "dendextend", "ComplexHeatmap", "rstatix",
"clusterProfiler", "DOSE", "org.Hs.eg.db", "EnhancedVolcano",
"GseaVis", "limma")
for (pkg in pkgs){
suppressPackageStartupMessages(library(pkg, character.only = T))
}
setwd("/cluster/home/yjliu_jh/projects/mm/output/response_diff")
# update gene name annotation
hugo_anno <- readr::read_delim("/cluster/home/yjliu_jh/projects/mm/data/hgnc_complete_set.txt",
col_types = cols(intermediate_filament_db = col_character()))
symbol_anno <- hugo_anno %>% separate_rows(prev_symbol, sep = "\\|") |> as.data.frame()
hugo_anno <- hugo_anno[, c("symbol", "ensembl_gene_id", "locus_group")] %>% as.data.frame()
colnames(hugo_anno)[2] <- "gene_id"
# load sampleinfo
merged_sinfo <- read_rds("~/projects/mm/docs/meta/sampleinfo/sampleinfo_jilin_commpass.rds")
resp_info <- merged_sinfo[, c(1:6, 30:33, 111, 143, 285:291)] %>% as.data.frame()
# select samples for comparison
resp_fil <- resp_info[!is.na(resp_info$subtypes), ]
resp_fil <- resp_fil[resp_fil$best_response %notin% "Unknown", ]
# get counts
counts_jilin <- read_csv("/cluster/home/jhuang/projects/mm/analysis/jilin/human/rnaseq/exp/tables/jilin_human_counts.csv")
counts_commpass <- read_csv("/cluster/home/jhuang/projects/mm/analysis/commpass/human/rnaseq/exp/tables/commpass_human_counts.csv")
counts_merged <- cbind(counts_jilin, counts_commpass[, -(1:2)])
counts_merged <- counts_merged[, -2] %>% left_join(hugo_anno) %>% na.omit() %>%
dplyr::select(-c(gene_id, locus_group)) %>% as.data.frame() %>% remove_rownames() %>%
column_to_rownames(var = "symbol") %>% as.matrix() %>% round()
counts_merged <- counts_merged[, resp_fil$sample_id]
#
rna_lib_large <- read_rds("/cluster/home/yjliu_jh/projects/mm/output/rna_lib_mm_samples.rds")
sinfo_comp <- left_join(resp_fil, rna_lib_large)
rownames(sinfo_comp) <- sinfo_comp$sample_id
sinfo_comp <- sinfo_comp[colnames(counts_merged), ]28 Treatment status in this study
Proteasome inhibitors and immunomodulatory agents are commonly used therapeutic drugs for multiple myeloma (MM). In our dataset, despite the heterogeneity in treatment combinations and regimens, the therapeutic strategies could be broadly categorized into four groups: (1) four-drug regimens based on daratumumab (DARA-based); (2) three-drug regimens combining proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs); (3) two-drug PI-based regimens; and (4) two-drug IMiD-based regimens. Among these, the three-drug and PI-based regimens were most prevalent, while the IMiD-based regimens were relatively rare. The DARA-based regimens were markedly underrepresented and were therefore excluded from the present analysis.
prepare data:
28.1 Treatment response groups and related functions
We applied consistent criteria to classify treatment responses in both the CoMMpass and Jilin datasets. Patients were grouped into seven response categories: stringent complete response (sCR), complete response (CR), very good partial response (VGPR), partial response (PR), minimal response (MR), stable disease (SD), and progressive disease (PD). For analysis, we designated sCR and CR as the best response group, and SD and PD as the poor response group, aiming to exclude ambiguous responses and maximize the contrast between the two groups for the identification of potentially response-associated biological functions.
sinfo_pionly_fil <- sinfo_pionly[sinfo_pionly$best_response %in% c("sCR", "CR", "SD", "PD"), ]
sinfo_pionly_fil$group <- ifelse(sinfo_pionly_fil$best_response %in% c("sCR", "CR"), "best", "worst")
counts_pionly_fil <- counts_merged[, sinfo_pionly_fil$sample_id]
dds_pi_part <- DESeqDataSetFromMatrix(counts_pionly_fil, sinfo_pionly_fil, ~ rna_type + group)
keep <- rowSums(counts(dds_pi_part) >= 5) > ceiling(0.2 * ncol(dds_pi_part))
dds_pi_part <- dds_pi_part[keep, ]
keep2 <- rowSums(counts(dds_pi_part) == 0) < ceiling(0.5 * ncol(dds_pi_part))
dds_pi_part <- dds_pi_part[keep2, ]
dds_pi_part <- DESeq(dds_pi_part)
res_pi_part <- add_col(results(dds_pi_part, c("group", "worst", "best")))
res_pi_part_fil <- as.data.frame(res_pi_part)[res_pi_part$pvalue < 0.05, ] %>% na.omit()
GSEAinput_pi2 <- res_pi_part$log2FoldChange
names(GSEAinput_pi2) <- as.character(rownames(res_pi_part))
GSEAinput_pi2 <- sort(GSEAinput_pi2, decreasing = TRUE)
gseGOres_pi2_part <- gseGO(GSEAinput_pi2, OrgDb = org.Hs.eg.db, keyType = "SYMBOL", ont = "BP",
minGSSize = 5, maxGSSize = 200, pvalueCutoff = 0.2)
# use batch-corrected exp data (limma)
mm1330 <- read_rds("~/projects/mm/analysis/meta/human/rnaseq/exp/mm1330.rds")
exp_mm <- mm1330[, -2] %>% left_join(hugo_anno) %>% na.omit() %>%
dplyr::select(-c(gene_id, locus_group)) %>% as.data.frame() %>% remove_rownames() %>%
column_to_rownames(var = "symbol") %>% as.matrix()
exp_pi_part <- exp_mm[, sinfo_pionly_fil$sample_id]
pi_group_part <- factor(sinfo_pionly_fil$group)
fit <- lmFit(exp_pi_part, model.matrix(~ pi_group_part))
fit <- eBayes(fit)
results_pi_part <- add_col2(topTable(fit, coef = 2, number = Inf, adjust.method = "BH"))
results_pi_part_fil <- results_pi_part[results_pi_part$P.Value < 0.05, ]
GSEA_input_pi_limma <- results_pi_part$logFC
names(GSEA_input_pi_limma) <- as.character(rownames(results_pi_part))
GSEA_input_pi_limma <- sort(GSEA_input_pi_limma, decreasing = TRUE)
gseGO_res_pi_part_limma <- gseGO(GSEA_input_pi_limma, OrgDb = org.Hs.eg.db, keyType = "SYMBOL", ont = "BP",
minGSSize = 5, maxGSSize = 200, pvalueCutoff = 0.2)Given the presence of batch effects and underlying subtype heterogeneity, we first assessed the impact of potential confounding factors by applying two complementary approaches: DESeq2 (correcting for sequencing platform only) and limma (applied to expression data with batch effects removed). Functional analysis of the resulting differential expression outputs revealed highly concordant results between the two methods, suggesting that batch effects and subtype differences had limited impact under these conditions.
ccres <- compareCluster(geneCluster = list(pi_deseq2 = GSEAinput_pi2, pi_limma = GSEA_input_pi_limma,
tri_deseq2 = GSEAinput_tri, tri_limma = GSEA_input_tri2_limma),
fun = gseGO, OrgDb = org.Hs.eg.db, keyType = "SYMBOL", ont = "BP",
minGSSize = 4, maxGSSize = 200, pvalueCutoff = 0.1)
pdf("four_comp.pdf", width = 8, height = 10)
dotplot(ccres)
dev.off()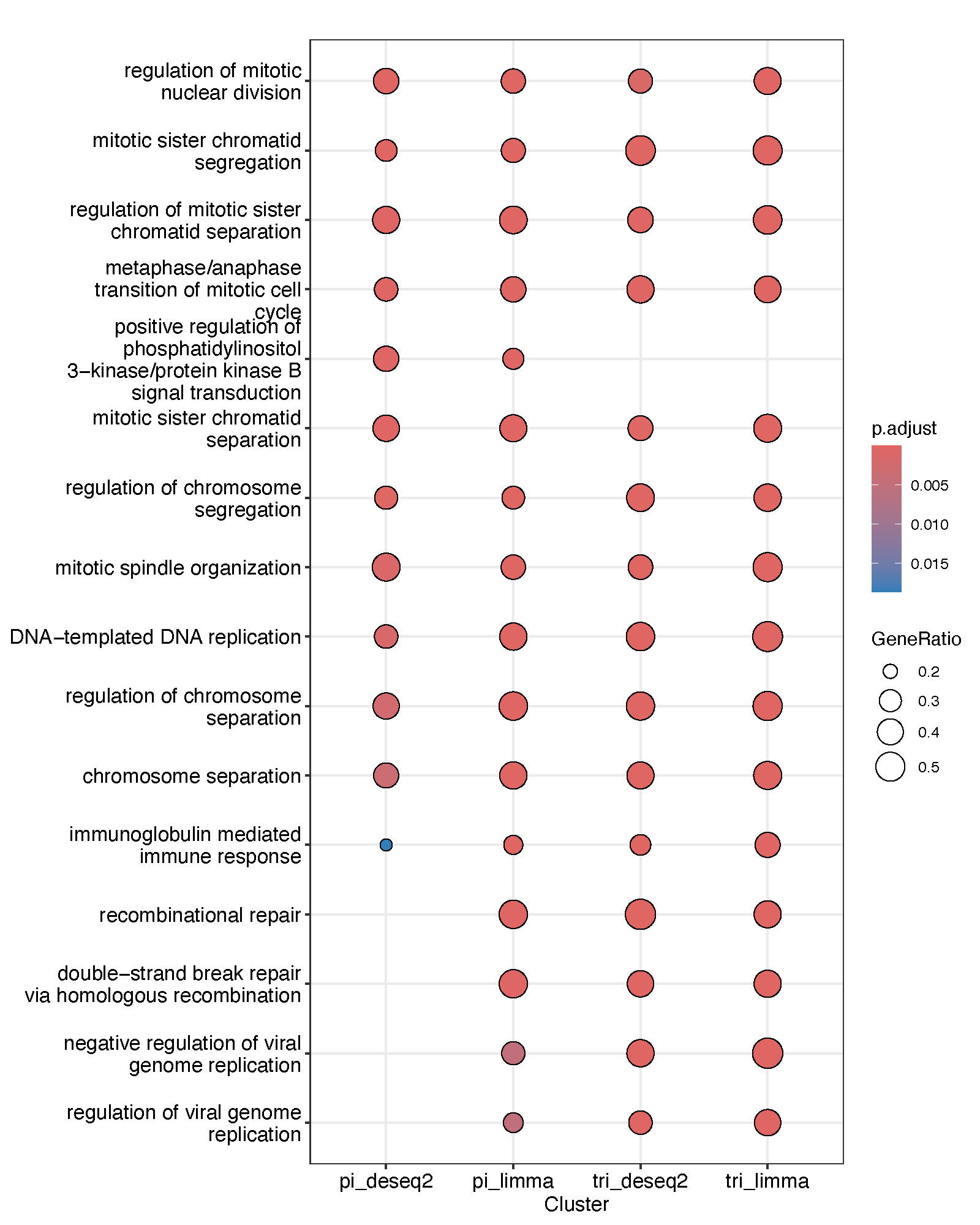
We next included the IMiD-based samples in our analysis and compared the resulting functional enrichment outcomes. Notably, sister chromatid separation consistently emerged as one of the most significantly enriched biological processes. This result was further validated using the Jilin dataset independently, indicating that this pathway exhibits robust differences across datasets and treatment regimens. Aberrations in sister chromatid separation can lead to aneuploidy, a hallmark of MM, and may offer insights into mechanisms of drug resistance.
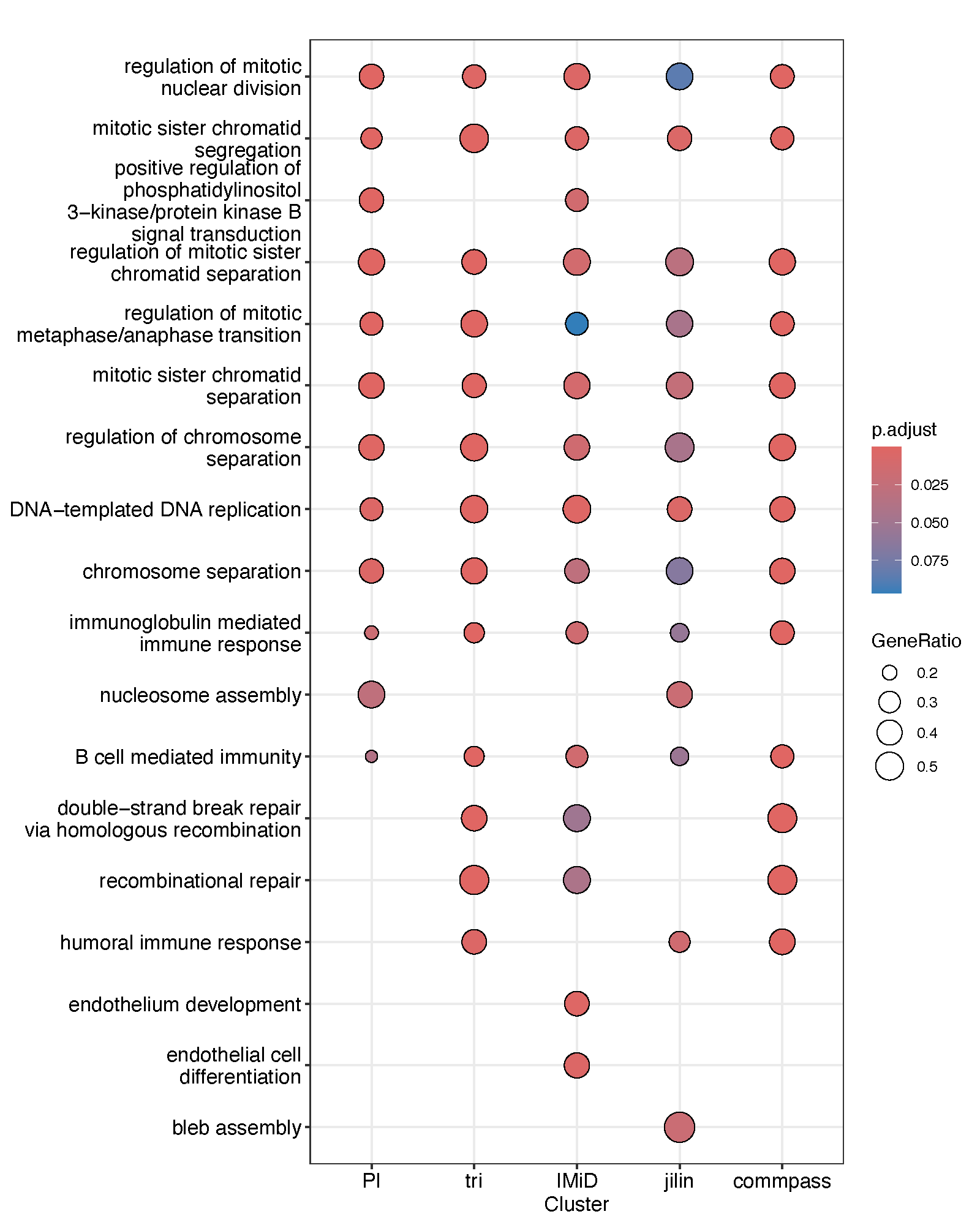
Subsequently, we investigated gene expression patterns associated with treatment response.
# match gene expression pattern to drug response groups
exp_resp <- exp_mm[, resp_fil$sample_id]
y7 <- resp_fil$group
trend_p <- apply(exp_resp, 1, \(g) cor.test(g, as.numeric(y7), method = "spearman")$p.value )
cand_genes <- names(head(sort(trend_p), 200))
# plot
resp_colors <- c("sCR" = "#F4E0FA", "CR" = "#E4E2F8", "VGPR" = "#9163C1", "PR" = "#F6D7ED",
"MR" = "#FEF2F2", "SD" = "#F7F2FE", "PD" = "#E0ECFA")
treat_colors <- RColorBrewer::brewer.pal(4, "Set1")
exp_long <- exp_resp %>% as.data.frame() %>% rownames_to_column("gene") %>%
pivot_longer(-gene, names_to = "sample_id", values_to = "expression")
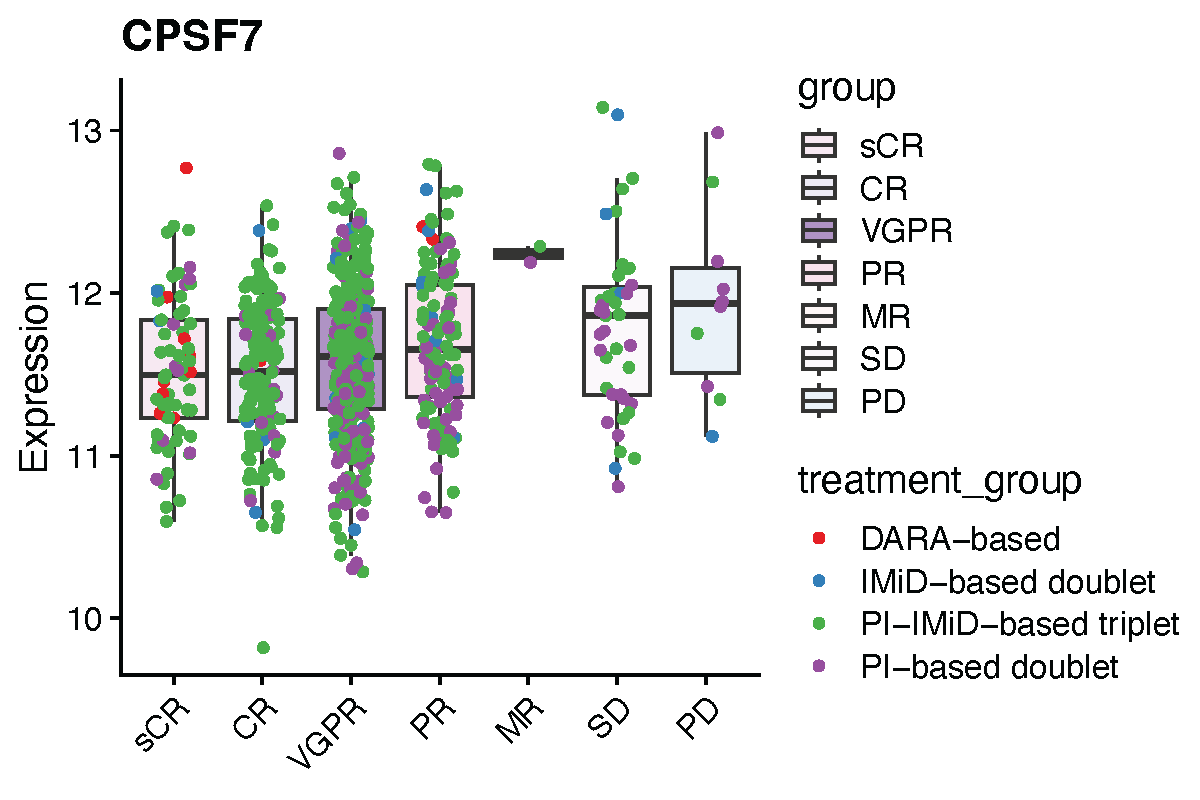
plot_data <- left_join(exp_long, resp_fil)No single gene was found to be strongly correlated with drug response. While certain genes showed modest expression trends corresponding to treatment efficacy, these patterns were insufficient to support predictive use. An example of such gene-level expression is illustrated below:
cand_gene <- "CPSF7"
gene_df <- plot_data[plot_data$gene %in% cand_gene, ]
p <- ggplot(gene_df, aes(x = group, y = expression)) +
geom_boxplot(aes(fill = group), outlier.shape = NA, alpha = 0.7) +
geom_jitter(aes(color = treatment_group), width = 0.2, size = 1.5) +
scale_fill_manual(values = resp_colors) +
scale_color_manual(values = treat_colors) +
labs(title = cand_gene, x = NULL, y = "Expression") +
theme_cowplot() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
ggsave(paste0("exp_plots/boxplot_", cand_gene, ".pdf"), plot = p, width = 6, height = 4)